Cluster Animation
Week 18





- Updated animation visuals to better emphasize the gene macropixel getting changed and the color change
- Adjusted the max number of bases in a sequence that can be changed by any one data input
- Testing complete program (animation and water system)
- Fixed screens going out of sync due to time delays between Arduino data input and animation pauses
- Plan to adjust initial main tank water level to be above middle level slightly (to ensure cyan dye does not drain before other behaviors can occur)
- Note that starting the animation program causes the Arduino to reset.
Week 17












- Running on 8-screen cluster
num_of_processes = 9- Randomized gene that gets updated for each data send
gene_rowid = random.randint(1, len(sequence_list)) #rank 0- Testing different animation background colors and room lighting for visibility (sticking with dark/neutral gray)
Week 16
Animation Composition Adjustment- Gene macropixel grid across full screen space (2x4 monitor set-up), centered, not subdivided per screen.
if rank != 0:
#...#
# create base pixel screen (separate surface)
base_genome_surface = pygame.Surface(universe_size)
base_genome_surface.fill(bg_color)
for row in range(universe_grid_y): # left to right, top down ordering of genes from dataset
for col in range(universe_grid_x):
color_id = color_id + 1
if color_id >= len(sequence_colors):
color_id = color_id_start
try:
pygame.draw.circle(base_genome_surface, sequence_colors[color_id], (upper_left_corner[0]+(pixel_width//2)+(pixel_width*col), upper_left_corner[1]+(pixel_height//2)+(pixel_height*row)), pixel_radius)
except:
print("Exception: color_id set out of bounds of sequence_colors at index", color_id)
# create screen/surface for showing just the updated element
update_gene_surface = pygame.Surface(universe_size)
update_gene_surface.fill(bg_color)- Periodic constant Arduino data sent by serial into animation, including tweaks for rate of sending due to difference between data sending speed and animation updating speed.
while True: # rank 0, code based on https://roboticsbackend.com/raspberry-pi-arduino-serial-communication/
if ser.in_waiting > 0: # according to the arduino sends, should occur at the same rate as the arduino timer (5s, maybe make greater)
line = ser.readline().decode('utf-8').rstrip()
print(line)
arduino_data = line.split(",") # ["#", "#", "#", "\n"]// from SafeString V3 library https://www.forward.com.au/pfod/ArduinoProgramming/TimingDelaysInArduino.html#examples
#include "loopTimer.h"
#include "millisDelay.h"
#include "BufferedOutput.h"
createBufferedOutput(bufferedOut, 80, DROP_UNTIL_EMPTY);
millisDelay printOutTimer;
void setup() {
Serial.begin(9600);
bufferedOut.connect(Serial);
printOutTimer.start(1000*10); // every 10 sec
}
void loop() {
bufferedOut.nextByteOut();
if(printOutTimer.justFinished()){
printOutTimer.repeat();
bufferedOut.print("400");
bufferedOut.print(",");
bufferedOut.print(220);
bufferedOut.print(",");
bufferedOut.print("600");
bufferedOut.print(",");
bufferedOut.println();
}
}Week 15
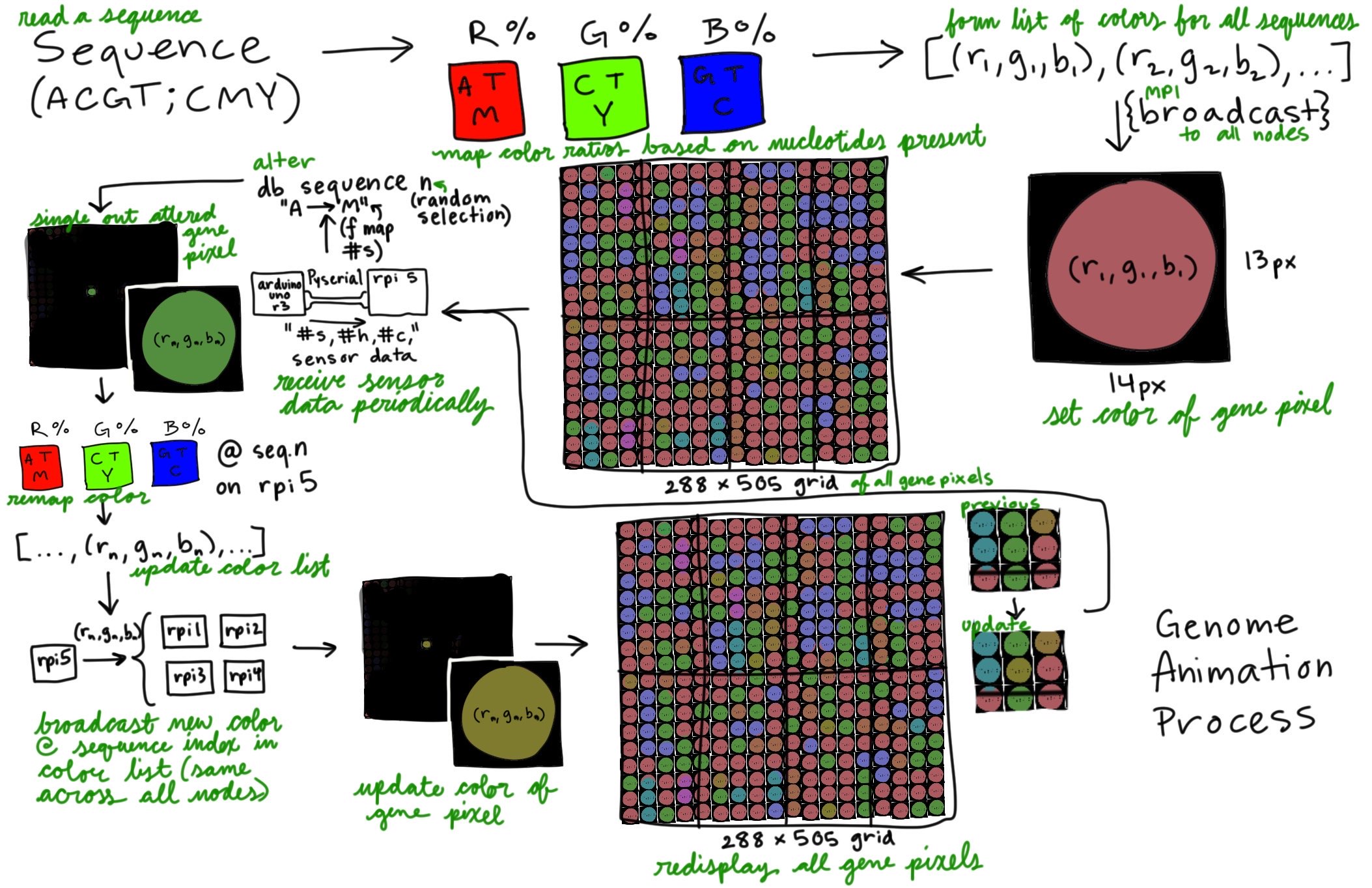


- Mapped sensor data ranges into colors and bases that directly replace bases in the given gene sequence of the genome data.
m_instances = int(np.interp(arduino_data[0], [0, 3000], [0, (len(gene_edit)-1)//3])) # number of bases to replace with M, direct map higher val = more bases
c_instances = int(np.interp(arduino_data[1], [120, 362], [0, (len(gene_edit)-1)//3])) # number of bases to replace with C, map lower water level w more bases (more dye added)
y_instances = int(np.interp(arduino_data[2], [0, 1023], [0, (len(gene_edit)-1)//3])) # number of bases to replace with Y, map only above threshold values; use if statement to make negatives into 0
print("# of bases to change m c y:",m_instances, c_instances, y_instances)
# get nonrepeating indices in sequence to change
replace_indices = random.sample(range(len(gene_edit)), k=(m_instances+c_instances+y_instances)) # gets nonrepeating set (equal to total bases to be replaced) of indices in sequence to change num
## https://stackoverflow.com/questions/1228299/changing-a-character-in-a-string/22149018#22149018 -> rec (for speed), splice a replacement in immutable string
# change the base at the specified index
for m in range(m_instances): # go over the first m_instances # of indices to replace, remove them each iteration to cont. use 0 index of sample list
gene_edit = gene_edit[:replace_indices[0]] + "M" + gene_edit[replace_indices[0]+1:] # splices around the index to change (i), and concatenates the new "base" in between
replace_indices.pop(0)
for c in range(c_instances): # go over the 1st(next) c_instances # of indices to replace, remove them each iteration to cont. use 0 index of sample list
gene_edit = gene_edit[:replace_indices[0]] + "N" + gene_edit[replace_indices[0]+1:] # splices around the index to change (i), and concatenates the new "base" in between
replace_indices.pop(0)
for y in range(y_instances): # go over the 1st(next) y_instances # of indices to replace, remove them each iteration to cont. use 0 index of sample list
gene_edit = gene_edit[:replace_indices[0]] + "Y" + gene_edit[replace_indices[0]+1:] # splices around the index to change (i), and concatenates the new "base" in between
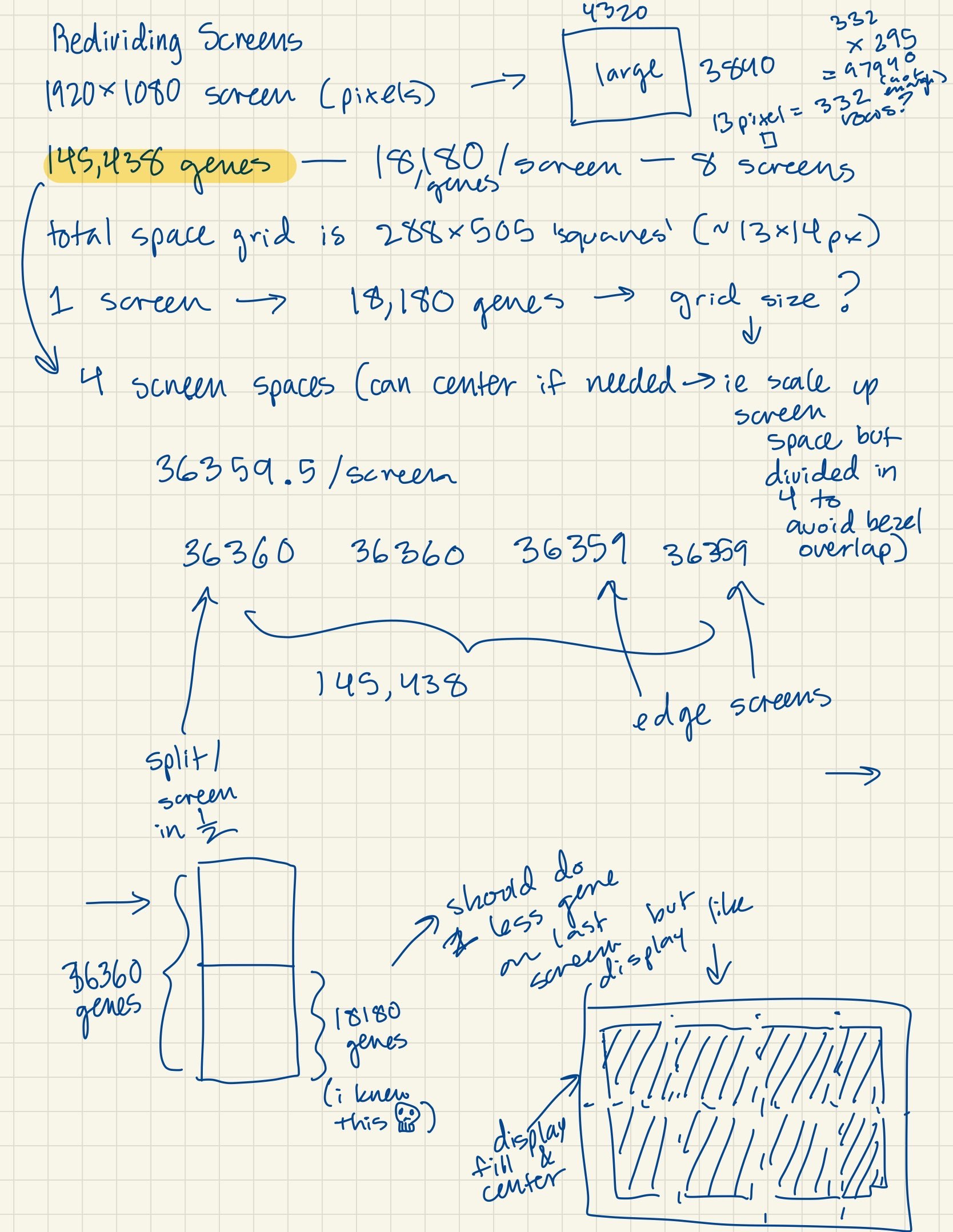
replace_indices.pop(0)- Divided portion of genes to each screen (approximately 18k gene macropixels per screen).
- Ideating how to divide genes continously across all screens (instead of assigning a specific subset to individual screens).
color_id_start = (grid_x*grid_y)*(rank-1) # set the index in the list of gene sequence colors for the given screen to start on (rank)
color_id = color_id_start- Scattered gene sequences across cluster nodes to process gene colors faster.
if rank == 0:
#.....#
num_of_processes = 4 # CHANGE WHEN PROGRAM IS ADJUSTED FOR NUM OF PROCESSES IN MPIRUN
split_gap = len(sequence_list) // num_of_processes
sequence_list_scatter = []
for i in range(num_of_processes): # 0 1 2 3 4 5 6 7 8
if i != (num_of_processes-1):
sequence_list_scatter.append(sequence_list[split_gap*i:(split_gap*i)+split_gap])
else:
sequence_list_scatter.append(sequence_list[split_gap*i:]) # to end
# scatter a 2d list of num_of_processes length
print("rank", rank, "waiting for sequence_list scatter")
sequence_list_scatter = comm.scatter(sequence_list_scatter, root=0)
print("rank:", rank, "received portion of sequence_list of length", len(sequence_list_scatter))- Using MPI broadcast function and sleep function to control speed of screen changes in animation.
print("new color:", sequence_colors[gene_rowid-1])
print("rank", rank, "sending new sequence_colors to ani")
comm.bcast(sequence_colors, root=0) # send out list again (could alternatively send out the sequence alone + rowid but idk if thats any faster)
# ^ at this point, the animation updates color for a moment on black screen still, then returns to normal full display; and rpi5 waits for new serial again
print("rank", rank, "sent new sequence_colors to ani")
- Animation running while excluding the primary node from showing animation.
- 8-screen cluster will have the primary node without a screen, so the command separates it from the animation and makes it only handle data processing.
>mpirun --host node1,node2:2,node3:2,node4:2 python3 gene_animation.pyWeek 13


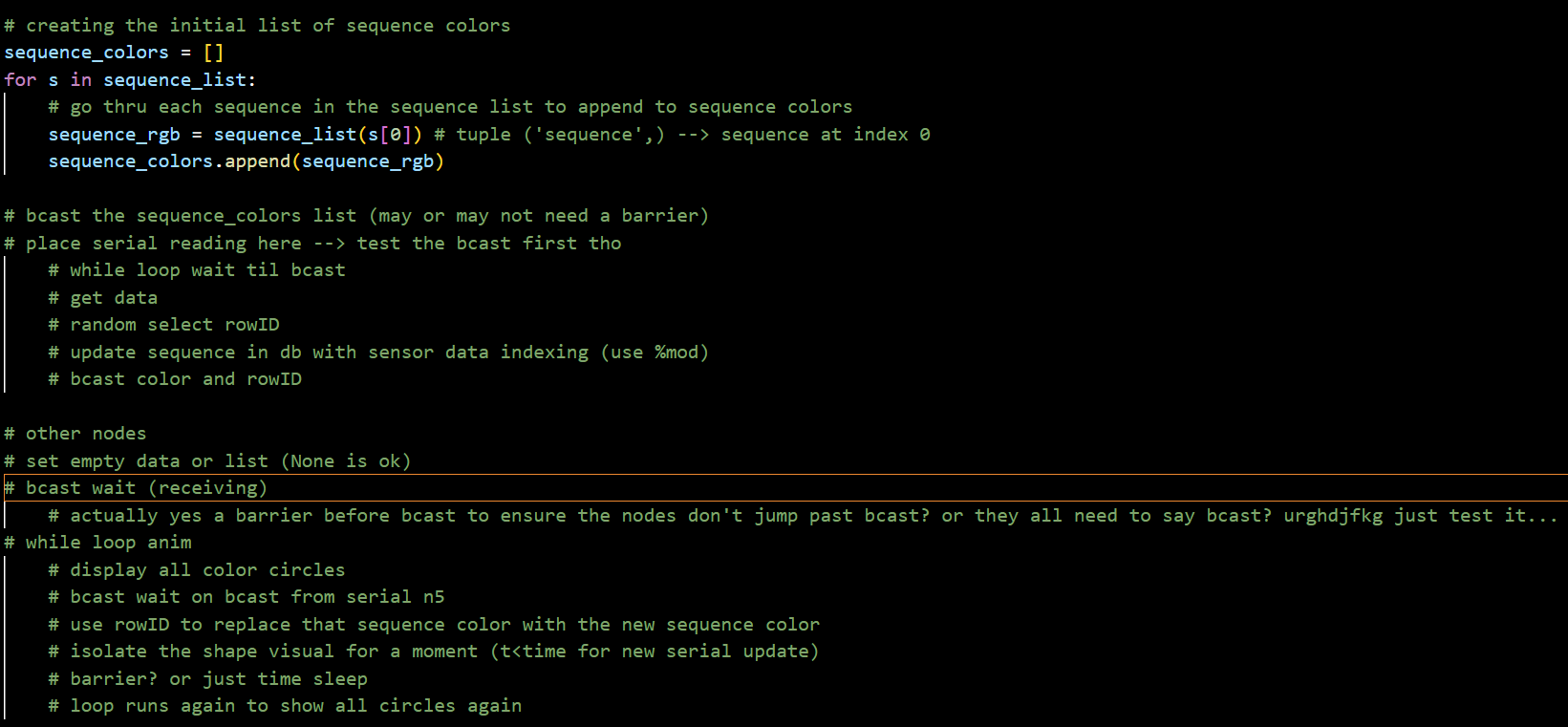


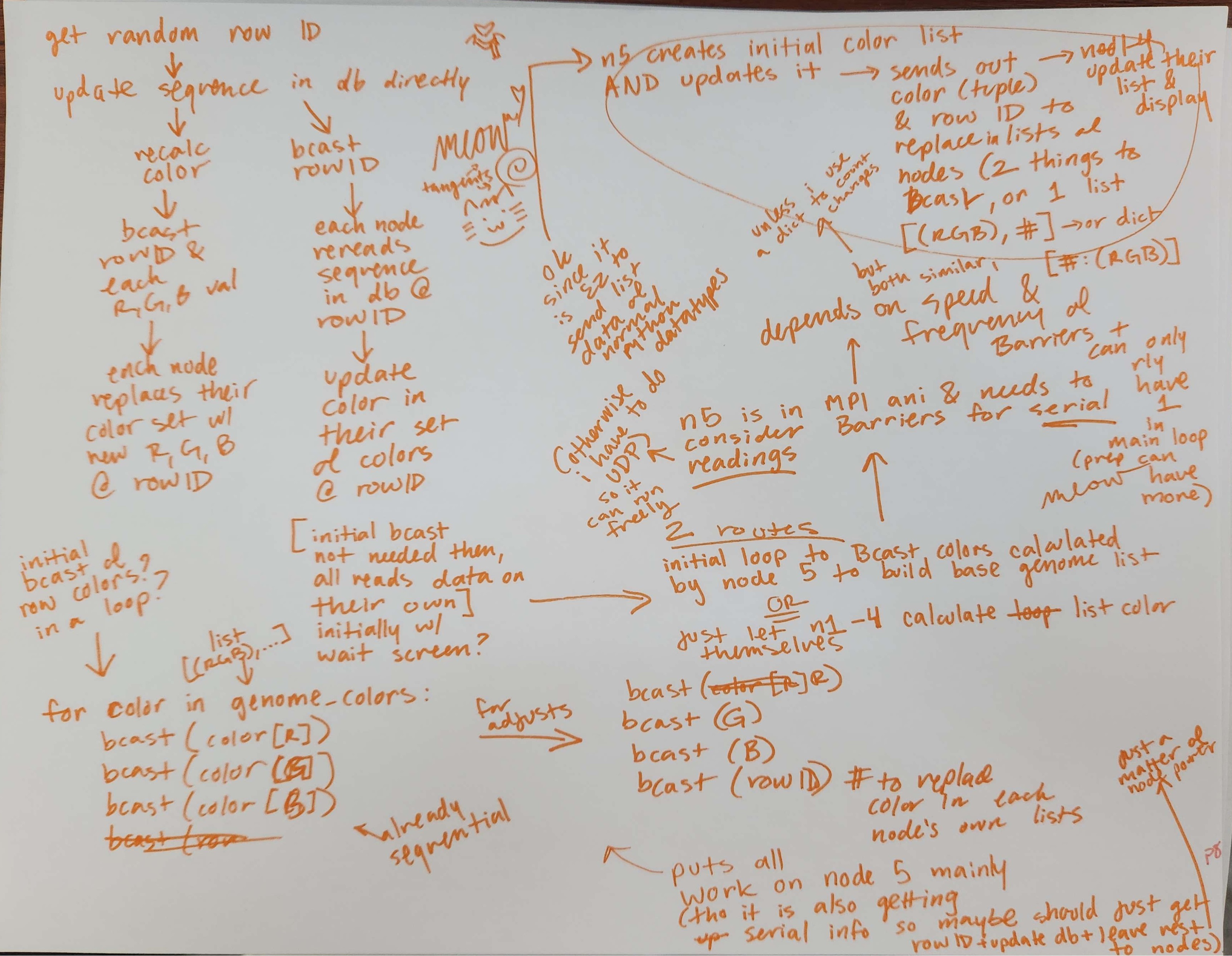
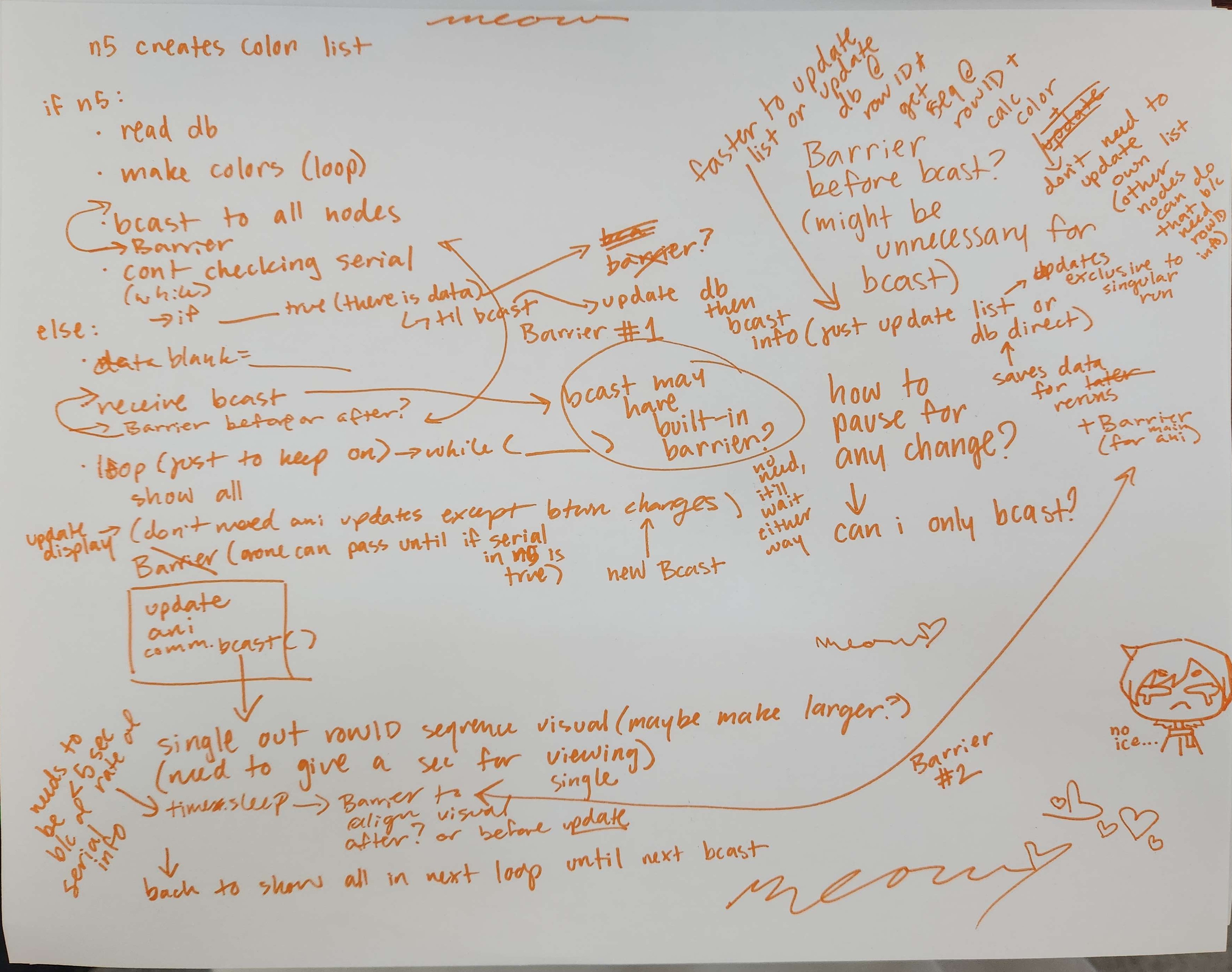
Tested broadcasting lists with MPI as potential format for distributing genome and Arduino data across all 5 computers.
Function for generating color of a gene sequence:
def sequence_color(sequence):
# generates the sequence color based on % of certain nucleotides in the sequence
len_sequence = len(sequence)
red_count = 0
green_count = 0
blue_count = 0
for n in sequence:
if n == "A":
red_count += 6
elif n == "C":
green_count += 6
elif n == "G":
blue_count += 6
elif n == "T":
red_count += 2
green_count += 2
blue_count += 2
elif n == "M":
red_count += 3
blue_count += 3
elif n == "N": # cyaN (since c is already used by genome)
green_count += 3
blue_count += 3
elif n == "Y":
red_count += 3
green_count += 3
else:
pass # not one of the bases, could be U? otherwise an error
# count of each nucleotide is worth 6 total so need to scale up sequence to 6 to get proper percentage i think..
# percentage of each color to be part of the RGB color of the overall sequence
red_per = red_count / (len_sequence*6)
green_per = green_count / (len_sequence*6)
blue_per = blue_count / (len_sequence*6)
# print(red_per, green_per, blue_per)
# actual color
red_val = 255*red_per //1
green_val = 255*green_per //1
blue_val = 255*blue_per //1
# return color
# print(red_val, green_val, blue_val)
return (red_val, green_val, blue_val)Week 12
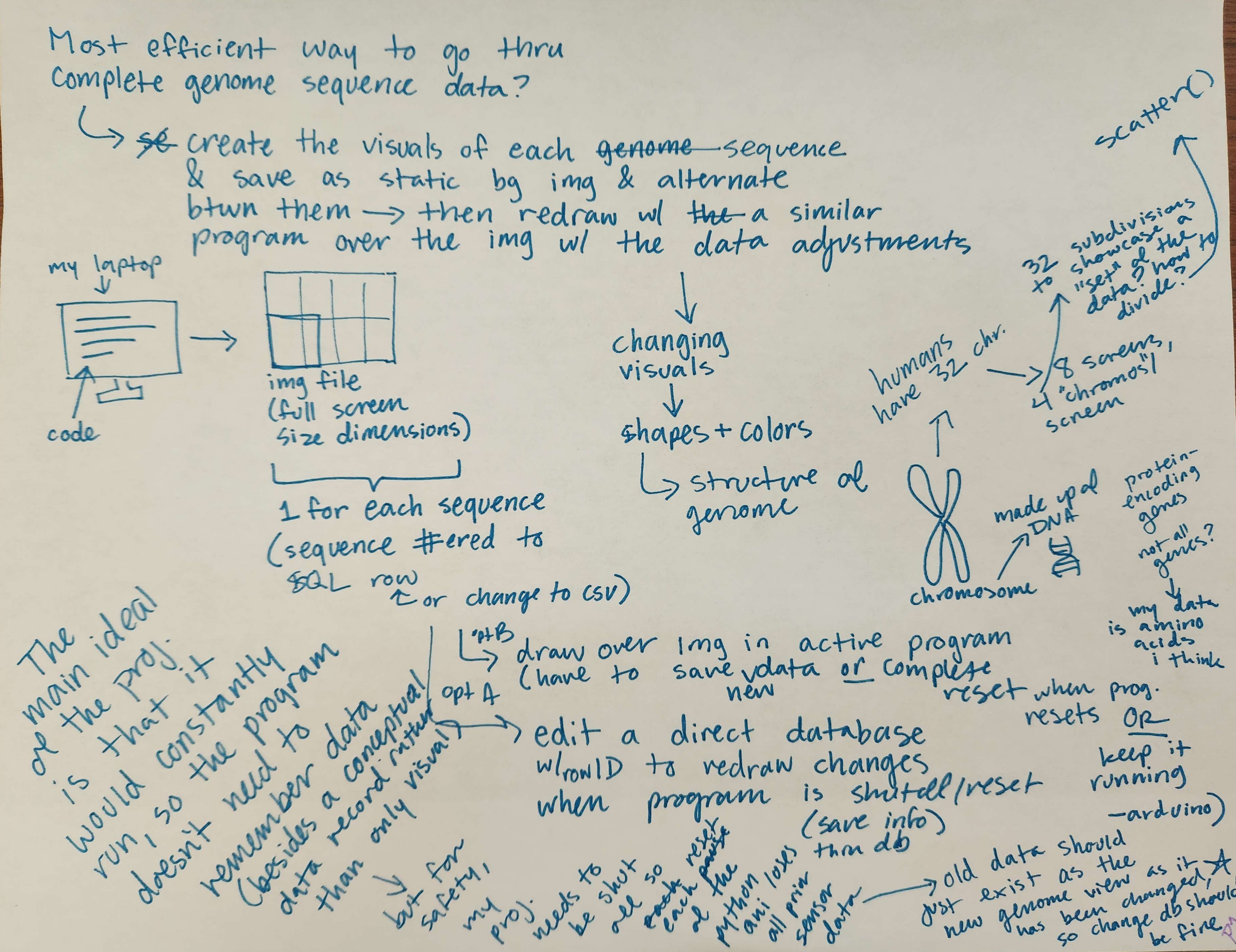



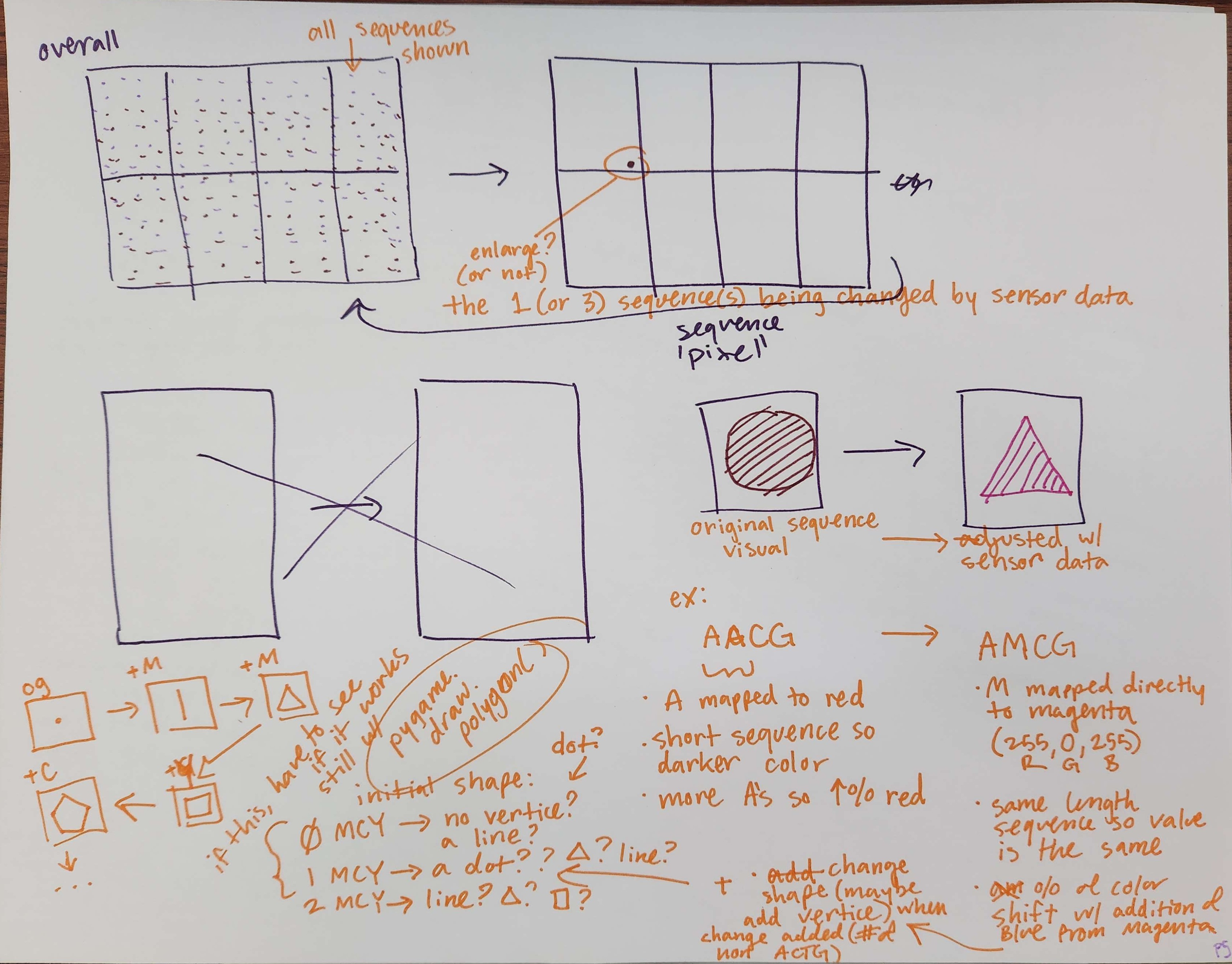
Concept notes on new animation visuals.
Week 11
Using PySerial to communicate Arduino's sensor data to Raspberry Pi program.
- Reference for PySerial: https://roboticsbackend.com/raspberry-pi-arduino-serial-communication/
Confirming that BufferedOutput library can be used to send data across serial port in place of Serial (Run PySerial program from roboticsbackend):
// from SafeString V3 library https://www.forward.com.au/pfod/ArduinoProgramming/TimingDelaysInArduino.html#examples
#include "loopTimer.h"
#include "millisDelay.h"
#include "BufferedOutput.h"
createBufferedOutput(bufferedOut, 80, DROP_UNTIL_EMPTY);
millisDelay printOutTimer;
void setup() {
Serial.begin(9600);
bufferedOut.connect(Serial);
printOutTimer.start(5000); // every 5 sec
}
void loop() {
bufferedOut.nextByteOut();
if(printOutTimer.justFinished()){ // print out the millis every 5 seconds
printOutTimer.repeat();
bufferedOut.println(millis());
}
}Send Arduino sensor data (through the global behavior variables) to the Raspberry Pi in the main code (snippet) [Removed extreme cases created in Week 10 due to errors making it impossible to properly test PySerial otherwise, see Week 10 Water Environment notes]:
void printData(){
if(printOutTimer.justFinished()){
printOutTimer.repeat();
bufferedOut.print(socialize);
bufferedOut.print(",");
bufferedOut.print(hydrate);
bufferedOut.print(",");
bufferedOut.print(create);
bufferedOut.print(",");
bufferedOut.println();
}
}
Week 9
WIP Genome Animation on 8-screen cluster set upimport os
import os.path
import sys
import pygame
from pygame.locals import *
import time
import random
from mpi4py import MPI
import math
import sqlite3
comm = MPI.COMM_WORLD
rank = comm.Get_rank()
size = comm.Get_size()
# set window position
os.environ['SDL_VIDEO_WINDOW_POS'] = "0,0"
# get this display
#print(dict(os.environ))
os.environ['DISPLAY'] = ':0.0'
# with caffeine installed (sudo apt install caffeine) we can wake the screen
# os.system('caffeinate sleep 1') # passes caffeinate out to shell to wake screensaver
# init clock and display
clock = pygame.time.Clock()
pygame.display.init()
pygame.mouse.set_visible(False)
# get the screen hight and width
disp_info = pygame.display.Info()
width = disp_info.current_w
height = disp_info.current_h
screen_size = (width,height) # adjust screen size manually is monitor is not the same dimensions as the main monitor
universe_size = (width*2, height*2)
screen = pygame.display.set_mode((screen_size), pygame.FULLSCREEN)
connection = sqlite3.connect('genome_human.db') # change to full path name?
cursor = connection.cursor()
cursor.execute("SELECT sequence FROM genome_human WHERE protein LIKE '%uncharacterized%'")
# SELECT * will return [(gene, protein, sequence),...]
# () <- tuple so even if 1 its still (value,) at least
# obtain thru tuple indexing: seq1 = var[0][0] (1st list item, index 0 of tuple)
var1 = cursor.fetchall()
connection.close()
def nucleotide_color(nucleotide):
if nucleotide == "A":
return (255,0,0) #red
elif nucleotide == "C":
return (0,255,0) #green
elif nucleotide == "T":
return (0,0,255) #blue
elif nucleotide == "G":
return (100,0,255) #purple?
else:
return (0,0,0)
def sine_wave(shift,sequence):
len_sequence = len(sequence)
for i in range(height):
# sine transformations -> bsin(ax-w)+h (b increases amplitude, a gives inverse frequency increase, w shifts horizontally, h shifts vertically)
# pygame.draw.line(screen,(50,100,255),(0,i),((width/2)+((width/2)*math.sin((height/4)*(i-shift))),i))
# moves down the screen
# pygame.draw.line(screen,(50,100,255),(0,i),((width/2)+((width/2)*math.sin((height/4)*i)),i))
# this (width/2)+((width/2)*math.sin((height/4)*i)) did NOT do what i expected at all but its GORGEOUS
if i >= len_sequence:
pygame.draw.line(screen,(255,255,255),(0,i),(int((width/2)+((width/2)*math.sin((height/4)*i))),i))
else:
pygame.draw.line(screen,nucleotide_color(sequence[i]),(0,i),(int((width/2)+((width/2)*math.sin((height/4)*i))),i))
return
framerate = 30
runtime = 10 #seconds
protein_index = 0
# time to run
# timeToRun = framerate*runtime
timeToRun = len(var1)
for f in range(timeToRun):
# screen.fill((50,30,255))
screen.fill((0,0,0))
sine_wave(f,var1[protein_index][0]) #shift later to go thru each sequence, this would be per frame but i could try other methods later
protein_index = protein_index + 1
if protein_index > len(var1):
protein_index = 0
comm.Barrier()
pygame.display.update()
clock.tick(framerate)Week 8
Preliminary animation of human genome uncharacterized proteins. Each frame is an individual sequence with the nucleotides mapped individually in color. (See code below)# from oneball.py (should also copy onto cluster) of VIS 141A
import os
import os.path
import sys
import pygame
from pygame.locals import *
import time
import random
# from mpi4py import MPI
import math
import sqlite3
# comm = MPI.COMM_WORLD
# rank = comm.Get_rank()
# size = comm.Get_size()
# set window position
os.environ['SDL_VIDEO_WINDOW_POS'] = "0,0"
# get this display
#print(dict(os.environ))
os.environ['DISPLAY'] = ':0.0'
# with caffeine installed (sudo apt install caffeine) we can wake the screen
# os.system('caffeinate sleep 1') # passes caffeinate out to shell to wake screensaver
# init clock and display
clock = pygame.time.Clock()
pygame.display.init()
pygame.mouse.set_visible(False)
# get the screen hight and width
disp_info = pygame.display.Info()
width = disp_info.current_w
height = disp_info.current_h
screen_size = (width,height) # adjust screen size manually is monitor is not the same dimensions as the main monitor
# universe_size = (width*2, height*2)
screen = pygame.display.set_mode((screen_size), pygame.NOFRAME)
# sqlite search cross with csv --> get list of uncharacterized protein codes, find each of those in sqlite database
# wondering if i should just put the protein names into sqlite db as well to better describe the protein and just search that way
# updated database: genome_human (gene VARCHAR(14), protein VARCHAR(132), sequence VARCHAR(107976), changed_sequence INTEGER)
# defaults changed_sequence to 0 (represents false), changes to 1 if serial data being read is giving into a change to the sequence (duplicate database or dataset to read)
connection = sqlite3.connect('genome_human.db') # change to full path name?
cursor = connection.cursor()
cursor.execute("SELECT sequence FROM genome_human WHERE protein LIKE '%uncharacterized%'")
# SELECT * will return [(gene, protein, sequence),...]
# () <- tuple so even if 1 its still (value,) at least
# obtain thru tuple indexing: seq1 = var[0][0] (1st list item, index 0 of tuple)
var1 = cursor.fetchall()
# cursor.execute("SELECT protein FROM genome_human WHERE protein LIKE 'uncharacterized%'")
# var2 = cursor.fetchall()
# print(len(var1)) #1638 (for protein)
# print(len(var2)) #1564 (for protein)
# check proteins that have augment before "uncharacterized"
# for p in var1:
# if p not in var2:
# print(p)
# described uncharacterized:
# ('LOW QUALITY PROTEIN: uncharacterized protein',)
# ('putative uncharacterized protein C3orf49',)
# do i want to include those above or even make them smth different based on being putative or low quality?
# print(var)
framerate = 24
runtime = 10 #seconds
def nucleotide_color(nucleotide):
if nucleotide == "A":
return 50
elif nucleotide == "C":
return 100
elif nucleotide == "T":
return 150
elif nucleotide == "G":
return 200
else:
return 255
def sine_wave(shift,sequence):
len_sequence = len(sequence)
for i in range(height):
# sine transformations -> bsin(ax-w)+h (b increases amplitude, a gives inverse frequency increase, w shifts horizontally, h shifts vertically)
# pygame.draw.line(screen,(50,100,255),(0,i),((width/2)+((width/2)*math.sin((height/4)*(i-shift))),i))
# moves down the screen
# pygame.draw.line(screen,(50,100,255),(0,i),((width/2)+((width/2)*math.sin((height/4)*i)),i))
# this (width/2)+((width/2)*math.sin((height/4)*i)) did NOT do what i expected at all but its GORGEOUS
if i >= len_sequence:
pygame.draw.line(screen,(255,255,255),(0,i),((width/2)+((width/2)*math.sin((height/4)*i)),i))
else:
pygame.draw.line(screen,(50,100,nucleotide_color(sequence[i])),(0,i),((width/2)+((width/2)*math.sin((height/4)*i)),i))
return
protein_index = 0
# time to run
# timeToRun = framerate*runtime
timeToRun = len(var1)
for f in range(timeToRun):
# screen.fill((50,30,255))
screen.fill((0,0,0))
sine_wave(f,var1[protein_index][0]) #shift later to go thru each sequence, this would be per frame but i could try other methods later
pygame.display.update()
protein_index = protein_index + 1
if protein_index > len(var1):
protein_index = 0
clock.tick(framerate)
connection.close()Week 7

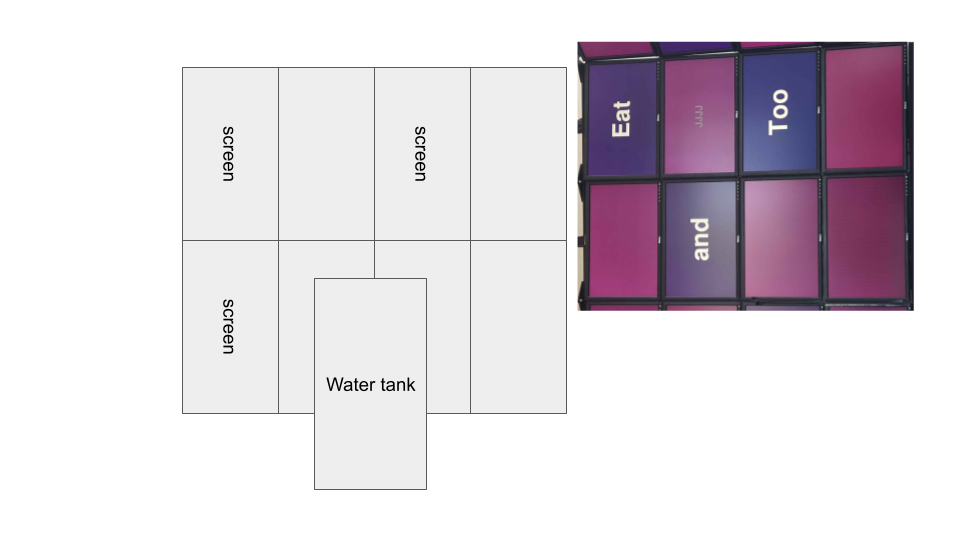
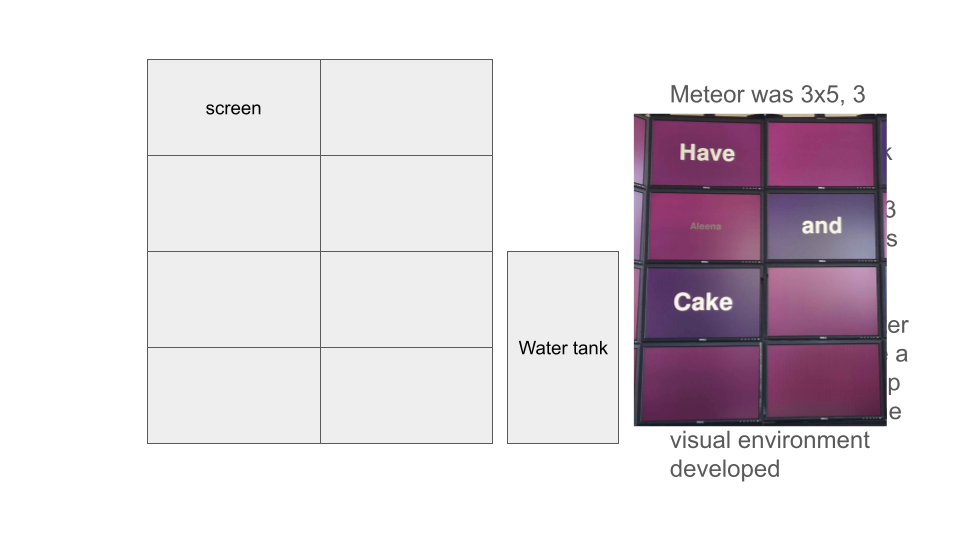
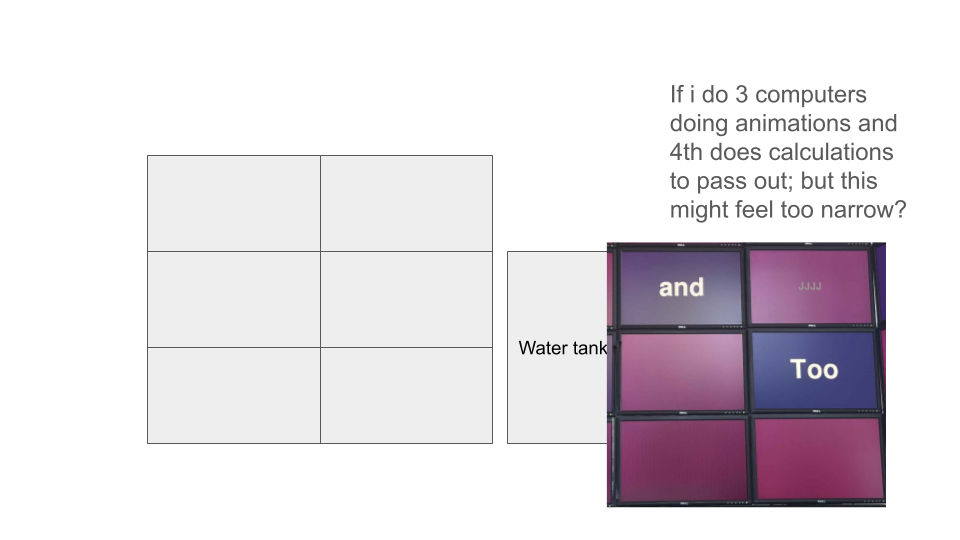

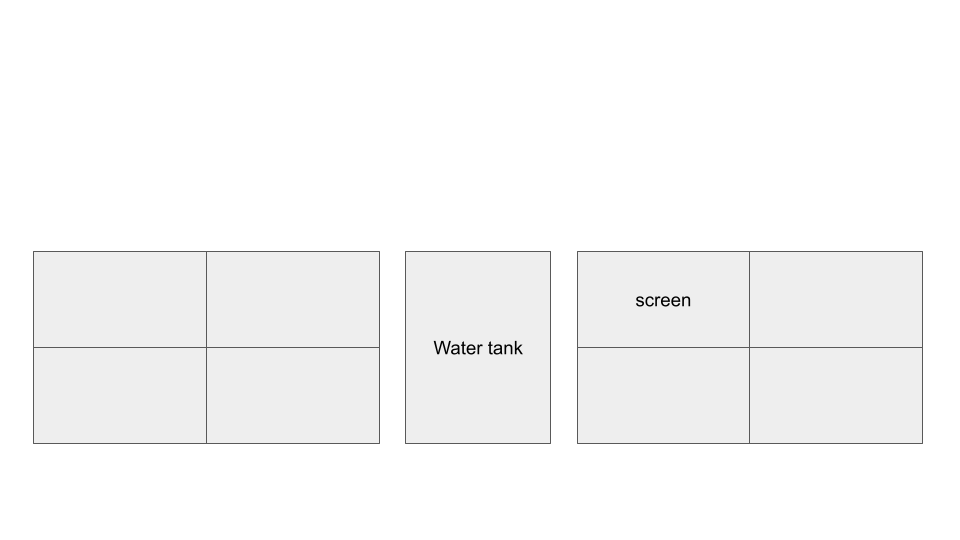
Rearranging screen display for cluster: 3x3 horizontal screens to 2x4 vertical screens. Intended to improve scaling and utilization of wall space.
Fixed genome .fna file reading: Wrote out gene ID, protein name, and sequence to SQLite database. (See code below)
Fixed PySerial communication and ran sample code to print out simple Serial output from Arduino:
# reading the human genome fna file
# code ref from ch 19 https://coding4medicine.com/backup/Python/reading-fasta-files.html
import sys
import time
import sqlite3
f=open("ncbi_dataset/data/GCF_000001405.40/cds_from_genomic.fna", "r") # open file with the genome data
connection = sqlite3.connect('genome_human.db') # change to full path name?
cursor = connection.cursor()
cursor.execute('CREATE TABLE genome_human (gene VARCHAR(14), protein VARCHAR(132), sequence VARCHAR(107976), changed_sequence INTEGER)') #determine the appropriate varchar max for each of these by reading through the file
# https://dba.stackexchange.com/questions/190674/can-a-database-table-not-have-a-primary-key
# use rowid to see primary key
# changed_sequence is boolean as integer (0 false, 1 true) --> indicates whether the sequence item has been changed or not by external adjustments to its nucleotides
connection.commit() # commit to creating the file before trying to insert into it
for line in f:
# read over each line of the file
if(line.startswith(">")):
if(new_gene):
new_gene = False
sequence_list = sequence_string.split("\n") # remove the \n from btwn each sequence subpart from the fna format and any blank lines
sequence_string = "" #clear out sequence so that it doesnt concatenate old sequences w new sequences
sequence_output = "".join(sequence_list) # join together that list to form one continuous sequence line
cursor.execute('INSERT INTO genome_human VALUES (?,?,?,0);', (gene_id, protein_name, sequence_output))
identity = line.split(" [") # split the elements of each into 1c1, gene], db], pro], proid], loc], gbkey] (since protein= can have spaces in btwn)
# gene always gene=###] --> same way to obtain as protein_id with splice or split and cut 5 (after =) to length
# protein_id will always be protein_id=#####] so can obtain number as split at = and get num minus ending bracket or splice index 11 to length (to keep out bracket)
# get gene
for i in identity:
if(i.startswith("gene=")):
gene_id = i
break
gene_id_len = len(gene_id)
gene_id = gene_id[5:(gene_id_len-1)] # cuts out "gene=" and "]" ; alternately can put len("gene=") in place of 6
for i in identity:
if(i.startswith("protein=")):
protein_name = i
break
protein_name_len = len(protein_name)
protein_name = protein_name[len("protein="):(protein_name_len-1)]
else:
new_gene = True
sequence_string = sequence_string + line # once starting a new gene, the string will be split at "\n" and pieced back together as a string
print("Done")
f.close()
connection.commit()
cursor.close()
connection.close()Week 5
Researching FASTA format of human genome data:
- https://coding4medicine.com/backup/Python/reading-fasta-files.html
- https://en.wikipedia.org/wiki/FASTA_format
- https://www.ncbi.nlm.nih.gov/datasets/taxonomy/9606/
Testing reading out genome data in Python:
- Issue: CSV file ended up being over 9GB!! (from a 2GB FASTA .fna file)
- Resolution: The variable holding each sequence was not being cleared for each new sequence and continuously concatenated the sequences (memory leak...).
Current issue: PySerial not installing on Raspberry Pi 5 (Spoiler Alert: It was already pre-installed)